Detecting signatures of selection with known environmental factors: a case of EnvGenomeScan
Xinghu Qin
7/11/2020
Source:vignettes/Detecting signatures of selection Simulation Case.Rmd
Detecting signatures of selection Simulation Case.RmdDeepGenomeScan for detection of natural selection with known environmental factors
In this vignette, we use the simulated data to show how to detect loci under the selection of known environmental factors using DeepGenomeScan. In this case, we use “EnvGenomeScan” mode in DeeGenomeScan, which does deep learning based genome scan to identify the loci explaining the environmental factors.
In what follows, we provide the step-by-step implementation of EnvGenomeScan.
Some examples run very long time, so we will not show the results of all examples, but only show examples runing quickly and shortly. Users can implement these methods on your own machine.
Load the required packages and dependencies
## Note: Install caret(plus) from xinghuq/CaretPlus/pkg/care #remotes::install_github("xinghuq/CaretPlus/pkg/caret") library("caret") # Loading required package: lattice # Loading required package: ggplot2 library("caretEnsemble") # # Attaching package: 'caretEnsemble' # The following object is masked from 'package:ggplot2': # # autoplot library("NeuralNetTools") library("neuralnet") library("DeepGenomeScan") # # Attaching package: 'DeepGenomeScan' # The following object is masked from 'package:caret': # # varImp library("ggplot2")
Get the example data.
Define the function to calculate the p-value and q-value
DLqvalues<-function(DL_data,K,estimation="auto") { require(robust) require(qvalue) loadings<-DL_data# [,1:as.numeric(K)] resscale <- apply(loadings, 2, scale) resmaha <- robust::covRob(resscale, distance = TRUE, na.action= na.omit, estim=estimation)$dist lambda <- median(resmaha)/qchisq(0.5,df=K) reschi2test <- stats::pchisq(resmaha/lambda,K,lower.tail=FALSE) qval <- qvalue::qvalue(reschi2test) q.values_DL<-qval$qvalues padj <- stats::p.adjust(reschi2test,method="bonferroni") return(data.frame(p.values=reschi2test, q.values=q.values_DL,padj=padj)) }
Machine learning models for genome scan
We will try different models to do genome scan. We will only show the step-by-step illustration, in which the tutorial will only search 10 hyperparameters here. Please do not take it for granted that these represent the best model performance. Please do a trial to explore the architecture of neural network that suits the data. Please increase the hyperparameter search space as well.
“mlpFCNN4Rsgd” example.
########## neuralnet mlp random search############################################ set.seed(123) econtrol <- caret::trainControl(## 5-fold CV, repeat 5 times method = "adaptive_cv", number = 5, ## repeated ten times repeats = 5, adaptive = list(min = 5, alpha = 0.05,method = "gls", complete = TRUE), search = "random" ) ###using FCNN4R #"sym_sigmoid" (and "tanh"), #library(FCNN4R) #### should install from source ##mlpFCNN4Rsgd mlpFCNN4Rsgd <- list(label = "Multilayer Perceptron Network by Stochastic Gradient Descent", library = c("FCNN4R", "plyr"), loop = NULL, type = c('Regression', "Classification"), parameters = data.frame(parameter = c('units1','units2', 'l2reg', 'lambda', "learn_rate", "momentum", "gamma", "minibatchsz", "repeats","activation1","activation2"), class = c(rep('numeric', 9), rep("character",2)), label = c('Number of hidden Units1','Number of hidden Units2', 'L2 Regularization', 'RMSE Gradient Scaling', "Learning Rate", "Momentum", "Learning Rate Decay", "Batch Size", "#Models",'Activation function at hidden layer','Activation function at output layer')), grid = function(x, y, len = NULL, search = "grid") { n <- nrow(x) afuncs=c("linear", "sigmoid", "tanh") if(search == "grid") { out <- expand.grid(units1 = ((1:len) * 2) - 1, units2 = ((1:len) * 2) - 1, l2reg = c(0, 10 ^ seq(-1, -4, length = len - 1)), lambda = 0, learn_rate = 2e-6, momentum = 0.9, gamma = seq(0, .9, length = len), minibatchsz = floor(nrow(x)/3), repeats = 1, activation1=c("linear", "sigmoid", "tanh"), activation2=c("linear", "sigmoid", "tanh")) } else { out <- data.frame(units1 = sample(2:20, replace = TRUE, size = len), units2 = sample(0:20, replace = TRUE, size = len), l2reg = 10^runif(len, min = -5, 1), lambda = runif(len, max = .4), learn_rate = runif(len), momentum = runif(len, min = .5), gamma = runif(len), minibatchsz = floor(n*runif(len, min = .1)), repeats = sample(1:10, replace = TRUE, size = len), activation1 = sample(afuncs, size = len, replace = TRUE), activation2 = sample(afuncs, size = len, replace = TRUE)) } out }, fit = function(x, y, wts, param, lev, last, classProbs, ...) { if(!is.matrix(x)) x <- as.matrix(x) if(is.factor(y)) { y <- class2ind(y) net <- FCNN4R::mlp_net(c(ncol(x), param$units1, param$units2,ncol(y))) net <- FCNN4R::mlp_set_activation(net, layer = "h", activation = param$activation1) net <- FCNN4R::mlp_set_activation(net, layer = "o", activation = param$activation2) } else { y <- matrix(y, ncol = 1) net <- FCNN4R::mlp_net(c(ncol(x), param$units1,param$units2, 1)) net <- FCNN4R::mlp_set_activation(net, layer = "h", activation = param$activation1) net <- FCNN4R::mlp_set_activation(net, layer = "o", activation =param$activation2) } n <- nrow(x) args <- list(net = net, input = x, output = y, learn_rate = param$learn_rate, minibatchsz = param$minibatchsz, l2reg = param$l2reg, lambda = param$lambda, gamma = param$gamma, momentum = param$momentum) the_dots <- list(...) if(!any(names(the_dots) == "tol_level")) { if(ncol(y) == 1) args$tol_level <- sd(y[,1])/sqrt(nrow(y)) else args$tol_level <- .001 } if(!any(names(the_dots) == "max_epochs")) args$max_epochs <- 1000 args <- c(args, the_dots) out <- list(models = vector(mode = "list", length = param$repeats)) for(i in 1:param$repeats) { args$net <- FCNN4R::mlp_rnd_weights(args$net) out$models[[i]] <- do.call(FCNN4R::mlp_teach_sgd, args) } out }, predict = function(modelFit, newdata, submodels = NULL) { if(!is.matrix(newdata)) newdata <- as.matrix(newdata) out <- lapply(modelFit$models, function(obj, newdata) FCNN4R::mlp_eval(obj$net, input = newdata), newdata = newdata) if(modelFit$problemType == "Classification") { out <- as.data.frame(do.call("rbind", out), stringsAsFactors = TRUE) out$sample <- rep(1:nrow(newdata), length(modelFit$models)) out <- plyr::ddply(out, plyr::`.`(sample), function(x) colMeans(x[, -ncol(x)]))[, -1] out <- modelFit$obsLevels[apply(out, 1, which.max)] } else { out <- if(length(out) == 1) out[[1]][,1] else { out <- do.call("cbind", out) out <- apply(out, 1, mean) } } out }, prob = function(modelFit, newdata, submodels = NULL) { if(!is.matrix(newdata)) newdata <- as.matrix(newdata) out <- lapply(modelFit$models, function(obj, newdata) FCNN4R::mlp_eval(obj$net, input = newdata), newdata = newdata) out <- as.data.frame(do.call("rbind", out), stringsAsFactors = TRUE) out$sample <- rep(1:nrow(newdata), length(modelFit$models)) out <- plyr::ddply(out, plyr::`.`(sample), function(x) colMeans(x[, -ncol(x)]))[, -1] out <- t(apply(out, 1, function(x) exp(x)/sum(exp(x)))) colnames(out) <- modelFit$obsLevels as.data.frame(out, stringsAsFactors = TRUE) }, varImp = function(object, ...) { imps <- lapply(object$models, caret:::GarsonWeights_FCNN4R, xnames = object$xNames) imps <- do.call("rbind", imps) imps <- apply(imps, 1, mean, na.rm = TRUE) imps <- data.frame(var = names(imps), imp = imps) imps <- plyr::ddply(imps, plyr::`.`(var), function(x) c(Overall = mean(x$imp))) rownames(imps) <- as.character(imps$var) imps$var <- NULL imps[object$xNames,,drop = FALSE] }, tags = c("Neural Network", "L2 Regularization"), sort = function(x) x[order(x$units1, x$units2,-x$l2reg, -x$gamma),]) varImpFCNN4 = function(object, ...) { imps <- lapply(object$models, caret:::GarsonWeights_FCNN4R, xnames = object$xNames) imps <- do.call("rbind", imps) imps <- apply(imps, 1, mean, na.rm = TRUE) imps <- data.frame(var = names(imps), imp = imps) imps <- plyr::ddply(imps, plyr::`.`(var), function(x) c(Overall = mean(x$imp))) rownames(imps) <- as.character(imps$var) imps$var <- NULL imps[object$xNames,,drop = FALSE] } for (i in 1:length(para)){ print(paste0("sim_1_",para[i]," train neuralnet")) Sys.time() #env=normalize(simdata[[j]][,2:11]) genotype_norm=as.data.frame(apply(sim_example,2,normalize)) simf=as.formula(paste(colnames(env)[i],paste(names(genotype_norm[,15:1014]), collapse="+"),sep="~")) model_FCNN4Rsgd_envi_mlp=DeepGenomeScan(simf, data=as.data.frame(genotype_norm), method="mlpSGD", metric = "MAE",## "Accuracy", "RMSE" #preProcess=c("scale"), tuneLength = 10, ### search 10 combinations of parameters # verbose=1 is reporting the progress,o is sclience trControl = econtrol) print(paste0("sim_1_",para[i],"tuning FCNN4Rsgd finished")) Sys.time() save(model_FCNN4Rsgd_envi_mlp,file=paste0("sim_1_",para[i],"_mlpFCNN4Rsgd_env_trained_model.RData")) ### olden importance # FCNN4Rsgd_simimp1=NeuralNetTools::olden(model_FCNN4Rsgd_envi_mlp$finalModel, bar_plot=FALSE) FCNN4Rsgd_simimp2=varImpFCNN4(model_FCNN4Rsgd_envi_mlp$finalModel) #write.csv(FCNN4Rsgd_simimp1$importance,file = paste0("sim_1_",para[i],"_mlpFCNN4Rsgd_importance_env.csv")) write.csv(FCNN4Rsgd_simimp2$Overall,file = paste0("sim_1_",para[i],"_mlpFCNN4Rsgd_importance2_env.csv")) write.csv(model_FCNN4Rsgd_envi_mlp$results,file = paste0("sim_1_",para[i],"_mlpFCNN4Rsgd_env_tuning.csv") ) print(paste0("sim_1_",para[i],"importance output finished")) #registerDoSEQ() #stopCluster(cl) } ##save.image(file = "FCNN4Rsgd_mlp_Scan_sims_final_after_trained_data.RData")
Reading the importance values and calculate p-values/q-values
impdir=list.dirs(path = "/home/xinghu2019/Simulations/DeepGenomeScan_v0.1/vignettes", full.names = TRUE, recursive = TRUE) impfile <- list.files(impdir, pattern="*_mlpFCNN4Rsgd_importance2_env.csv", full.names = TRUE) ## 100 simulations and 10 envir variables, impSim_all=lapply(impfile, read.csv) ### including all 100 simulations and 10 variables each simuilations, ### combine every factor (envir) into one file, meaning combining each single simulation into one file imp_sim_list=as.data.frame(do.call(cbind, impSim_all)) ### remove the loci ID Xrep<- !names(imp_sim_list) %in% c("X") DLdata <- imp_sim_list[,Xrep, drop = FALSE] colnames(DLdata) ### now 1:10 is the first simulation ### spilt into 100 simulations each by 10 #save.image(file = "DL_mlp_FNCC4_Scan_sim1.RData") ###calculate q-values DLdata1=apply(DLdata,2,normalize) Simqvalue=DLqvalues(DLdata1,10) # Loading required package: robust # Loading required package: fit.models # Loading required package: qvalue Loci<-rep("Neutral", 1000) Loci[c(1,11,21,31,41,51,61,71,81,91)]<-"QTL1" Loci[c(101,111,121,131,141,151,161,171,181,191)]<-"QTL2" Loci[c(201,211,221,231,241,251,261,271,281,291)]<-"QTL3" p.values<-data.frame(index = c(1:1000), DL_mlp=-log10(Simqvalue$p.values),Loci=Loci) Selected_Loci<-p.values$Loci[-which(p.values$Loci=="Neutral")] # [1] "x" "x.1" "x.2" "x.3" "x.4" "x.5" "x.6" "x.7" "x.8" "x.9"
Plotting Manhattan plot
load("DL_mlp_FNCC4_Scan_sim1_manhattan.RData") p1 <- ggplot() + geom_point(aes(x=p.values$index[-which(p.values$Loci!="Neutral")], y=p.values$DL_mlp[-which(p.values$Loci!="Neutral")]), col = "gray83") + geom_point(aes(x=as.vector(p.values$index[-which(p.values$Loci=="Neutral")]), y=as.vector(p.values$DL_mlp[-which(p.values$Loci=="Neutral")]), colour = Selected_Loci)) + xlab("SNP (with maf > 0.05)") + ylab("DL_MLP -log10(p-values)") + ylim(0,120) + theme_bw() p1
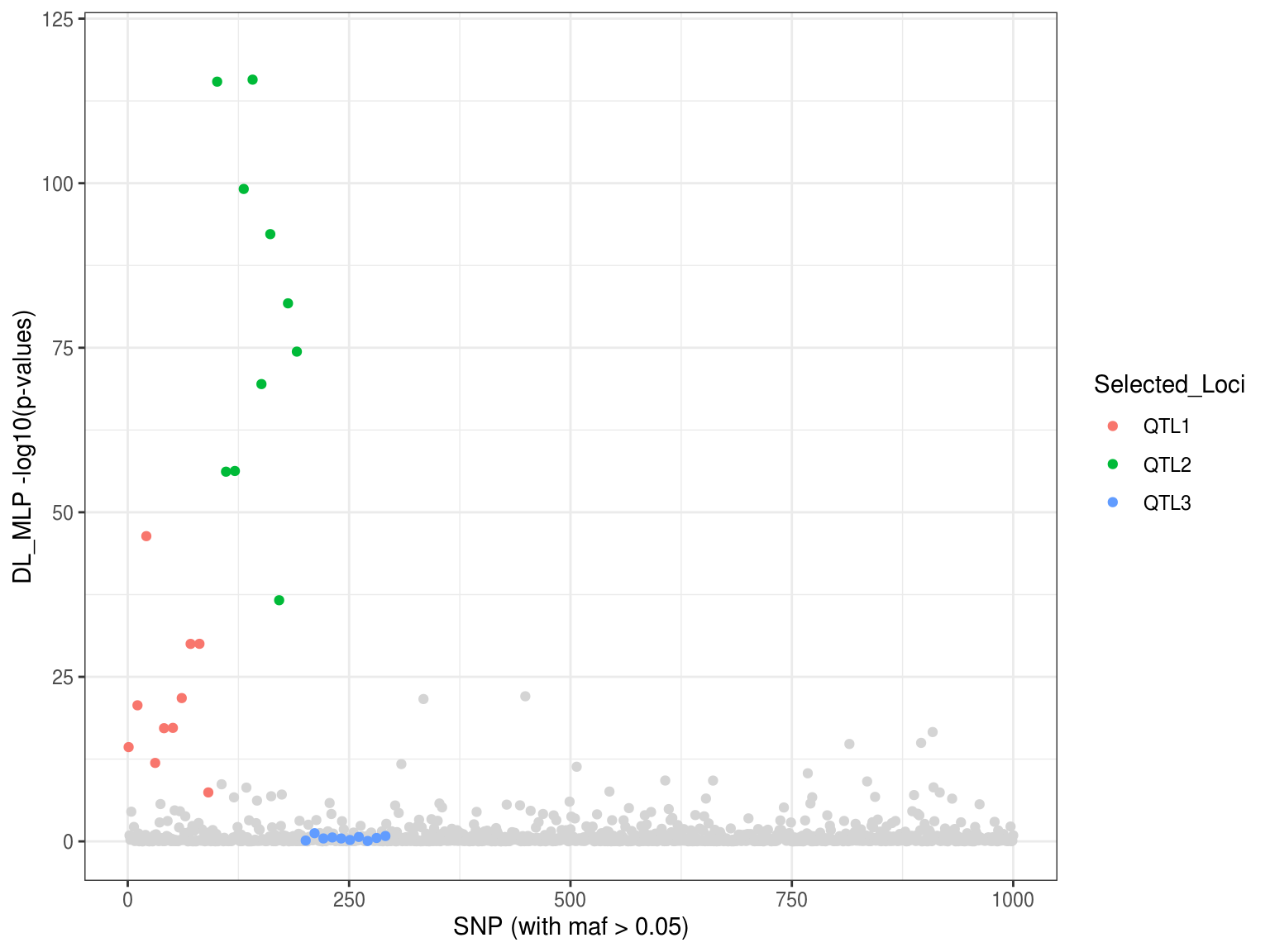
Doing genome scan with the compiled model.
library(h2o) set.seed(123) econtrol <- caret::trainControl(## 5-fold CV, repeat 5 times method = "adaptive_cv", number = 5, ## repeated ten times repeats = 5, adaptive = list(min = 5, alpha = 0.05,method = "gls", complete = TRUE), search = "random") for (i in 1:length(env)){ print(paste0("sim_1_",para[i]," train neuralnet")) Sys.time() localH2o <- h2o.init(nthreads = -1, max_mem_size = "6G") h2o.no_progress() #env=normalize(simdata[[j]][,2:11]) genotype_norm=as.data.frame(apply(genotype,2,normalize)) #simf=as.formula(paste(colnames(env)[i],paste(names(genotype_norm[,15:1014]), collapse="+"),sep="~")) model_mlph2o_envi_mlp=DeepGenomeScan(genotype=genotype_norm,env=env[,i], method=DL_h2o, metric = "MAE",## "Accuracy", "RMSE" #preProcess=c("scale"), tuneLength = 10, ### search 10 combinations of parameters verbose=0, #is reporting the progress,o is sclience trControl = econtrol) print(paste0("sim_1_",para[i],"tuning mlph2o finished")) Sys.time() save(model_mlph2o_envi_mlp,file=paste0("sim_1_",para[i],"_mlph2o_env_trained_model.RData")) ### olden importance # mlph2o_simimp1=NeuralNetTools::olden(model_mlph2o_envi_mlp$finalModel, bar_plot=FALSE) mlph2o_simimp2=as.data.frame(h2o::h2o.varimp(model_mlph2o_envi_mlp$finalModel)) #write.csv(mlph2o_simimp1$importance,file = paste0("sim_1_",para[i],"_mlpmlph2o_importance_env.csv")) ####note: rearrange the order of row locname=colnames(genotype) macha=match(locname,mlph2o_simimp2$variable) ragimp=mlph2o_simimp2[macha,] write.csv(ragimp$scaled_importance,file = paste0("sim_1_",para[i],"_mlph2o_importance2_env.csv")) write.csv(model_mlph2o_envi_mlp$results,file = paste0("sim_1_",para[i],"_mlph2o_env_tuning.csv") ) print(paste0("sim_1_",para[i],"importance output finished")) #registerDoSEQ() #stopCluster(cl) } ##save.image(file = "mlph2o_mlp_Scan_sims1_final_after_trained_data.RData") ## Defining function to calculate the p-value and q-values DLqvalues<-function(DL_data,K,estimation="auto") { require(robust) require(qvalue) loadings<-DL_data# [,1:as.numeric(K)] resscale <- apply(loadings, 2, scale) resmaha <- robust::covRob(resscale, distance = TRUE, na.action= na.omit, estim=estimation)$dist lambda <- median(resmaha)/qchisq(0.5,df=K) reschi2test <- stats::pchisq(resmaha/lambda,K,lower.tail=FALSE) qval <- qvalue::qvalue(reschi2test) q.values_DL<-qval$qvalues padj <- stats::p.adjust(reschi2test,method="bonferroni") return(data.frame(p.values=reschi2test, q.values=q.values_DL,padj=padj)) }
Reading importance values and calculate the p-values/q-values
impdir=list.dirs(path = ".", full.names = TRUE, recursive = TRUE) impfile <- list.files(impdir, pattern="*_mlph2o_importance_update_env.csv", full.names = TRUE) ## 100 simulations and 10 envir variables, impSim_all=lapply(impfile, read.csv) ### including all 100 simulations and 10 variables each simuilations, ### combine every factor (envir) into one file, meaning combining each single simulation into one file imp_sim_list=as.data.frame(do.call(cbind, impSim_all)) ### remove the loci ID Xrep<- !names(imp_sim_list) %in% c("X") DLdata <- imp_sim_list[,Xrep, drop = FALSE] colnames(DLdata) ### now 1:10 is the first simulation ### spilt into 100 simulations each by 10 save.image(file = "DL_mlp_h2o_Scan_sim1.RData") ###calculate q-values DLdata1=apply(DLdata,2,normalize) Simqvalue=DLqvalues(DLdata1,10) Loci<-rep("Neutral", 1000) Loci[c(1,11,21,31,41,51,61,71,81,91)]<-"QTL1" Loci[c(101,111,121,131,141,151,161,171,181,191)]<-"QTL2" Loci[c(201,211,221,231,241,251,261,271,281,291)]<-"QTL3" p.values<-data.frame(index = c(1:1000), DL_mlp=-log10(Simqvalue$p.values),Loci=Loci) Selected_Loci<-p.values$Loci[-which(p.values$Loci=="Neutral")]
Plotting Manhattan plot
load(file = "DL_mlph2o_Scan_sim1_manhattan.RData") p2 <- ggplot() + geom_point(aes(x=p.values$index[-which(p.values$Loci!="Neutral")], y=p.values$DL_mlp[-which(p.values$Loci!="Neutral")]), col = "gray83") + geom_point(aes(x=as.vector(p.values$index[-which(p.values$Loci=="Neutral")]), y=as.vector(p.values$DL_mlp[-which(p.values$Loci=="Neutral")]), colour = Selected_Loci)) + xlab("SNP (with maf > 0.05)") + ylab("DL_MLP -log10(p-values)") + ylim(0,40) + theme_bw() p2
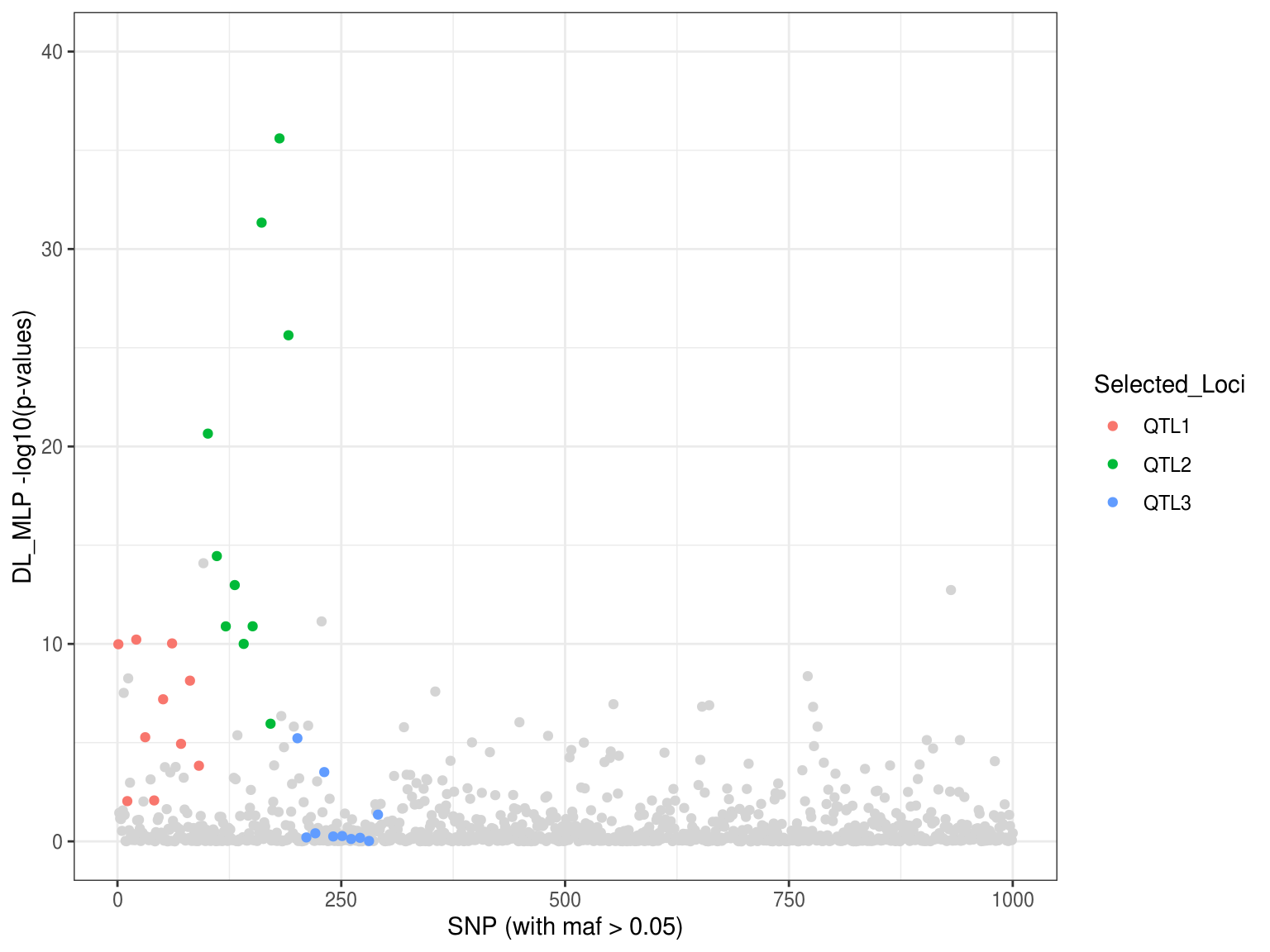
#save.image(file = "DL_mlph2o_Scan_sim1_manhattan.RData")h2o already has its own framework that can train, tune, and optimize the model. Let’s firt implements h2o’s own framework.
Reading importance values and calculate the p-values/q-values
impdir=list.dirs(path = ".", full.names = TRUE, recursive = TRUE) impfile <- list.files(impdir, pattern="*_mlph2o_varimp_env.csv", full.names = TRUE) ## 100 simulations and 10 envir variables, impSim_all=lapply(impfile, read.csv) ### including all 100 simulations and 10 variables each simuilations, ### combine every factor (envir) into one file, meaning combining each single simulation into one file imp_sim_list=as.data.frame(do.call(cbind, impSim_all)) ### remove the loci ID Xrep<- !names(imp_sim_list) %in% c("X") DLdata <- imp_sim_list[,Xrep, drop = FALSE] colnames(DLdata) ### now 1:10 is the first simulation ### spilt into 100 simulations each by 10 save.image(file = "DL_mlp_pureh2o_Scan_sim1.RData") ###calculate q-values DLdata1=apply(DLdata,2,normalize) Simqvalue=DLqvalues(DLdata,10) Loci<-rep("Neutral", 1000) Loci[c(1,11,21,31,41,51,61,71,81,91)]<-"QTL1" Loci[c(101,111,121,131,141,151,161,171,181,191)]<-"QTL2" Loci[c(201,211,221,231,241,251,261,271,281,291)]<-"QTL3" p.values<-data.frame(index = c(1:1000), DL_mlp=-log10(Simqvalue$q.values),Loci=Loci) Selected_Loci<-p.values$Loci[-which(p.values$Loci=="Neutral")]
Plotting Manhattan plot
load("DL_mlph2o_Scan_sim1_manhattan.RData") p3 <- ggplot() + geom_point(aes(x=p.values$index[-which(p.values$Loci!="Neutral")], y=p.values$DL_mlp[-which(p.values$Loci!="Neutral")]), col = "gray83") + geom_point(aes(x=as.vector(p.values$index[-which(p.values$Loci=="Neutral")]), y=as.vector(p.values$DL_mlp[-which(p.values$Loci=="Neutral")]), colour = Selected_Loci)) + xlab("SNP (with maf > 0.05)") + ylab("DL_MLP -log10(p-values)") + ylim(0,40) + theme_bw() p3 #save.image(file = "DL_mlph2o_Scan_sim1_manhattan.RData")
Neural network example with “nnet”
Now we use nnet library to construct the neural netwok.
econtrol <- caret::trainControl( ## 5-fold CV, repeat 5 times method = "adaptive_cv", number = 5, ## repeated ten times repeats = 5, adaptive = list(min = 5, alpha = 0.05,method = "gls", complete = TRUE),search = "random") for (i in 1:length(para)){ print(paste0("sim_1_",para[i],"tuning nnet tree 10 parameters")) Sys.time() #env=normalize(simdata[[j]][,2:11]) #genotype_norm=as.data.frame(apply(simdata,2,normalize)) # simf=as.formula(paste(colnames(env)[i],paste(names(genotype_norm[,15:1014]), collapse="+"),sep="~")) model_nnet_envi=DeepGenomeScan(genotype=genotype_norm, env=env[,i], method="nnet", metric = "MAE",## "Accuracy", "RMSE" #preProcess=c("scale"), tuneLength = 10, ### search 10 combinations of parameters # verbose=1 is reporting the progress,o is sclience trControl = econtrol,maxit=100,MaxNWts=94581) print(paste0("sim_",1,"_",para[i],"tuning nnet finished")) Sys.time() save(model_nnet_envi,file=paste0("sim_",1,"_",para[i],"_nnet_Scan_env_trained_model.RData")) ### olden importance nnet_simimp1=varImp(model_nnet_envi$finalModel) nnet_oldenimp=NeuralNetTools::olden(model_nnet_envi$finalModel,bar_plot=FALSE) write.csv(nnet_simimp1$Overall,file = paste0("sim_",1,"_",para[i],"_nnet_Scan_importance_env.csv")) write.csv(nnet_oldenimp$importance,file = paste0("sim_",1,"_",para[i],"_nnet_Scan_oldenimportance_env.csv")) write.csv(model_nnet_envi$results,file = paste0("sim_",1,"_",para[i],"_model_nnet_envi_tuning.csv") ) #registerDoSEQ() #stopCluster(cl) }
Defining the function to calculate the Mahalanobis distance.
DLqvalues<-function(DL_data,K,estimation="donostah") { require(robust) require(qvalue) loadings<-DL_data# [,1:as.numeric(K)] resscale <- apply(loadings, 2, scale) resmaha <- robust::covRob(resscale, distance = TRUE, na.action= na.omit, estim=estimation)$dist lambda <- median(resmaha)/qchisq(0.5,df=K) reschi2test <- stats::pchisq(resmaha/lambda,K,lower.tail=FALSE) qval <- qvalue::qvalue(reschi2test) q.values_DL<-qval$qvalues padj <- stats::p.adjust(reschi2test,method="bonferroni") return(data.frame(p.values=reschi2test, q.values=q.values_DL,padj=padj)) }
Calculating the Mahalanobis distance and p-values/q-values
impdir=list.dirs(path = ".", full.names = TRUE, recursive = TRUE) impfile <- list.files(impdir, pattern="*_nnet_Scan_oldenimportance_env.csv", full.names = TRUE) ## 100 simulations and 10 envir variables, impSim_all=lapply(impfile, read.csv) ### including all 100 simulations and 10 variables each simuilations, ### combine every factor (envir) into one file, meaning combining each single simulation into one file imp_sim_list=as.data.frame(do.call(cbind, impSim_all)) ### remove the loci ID Xrep<- !names(imp_sim_list) %in% c("X") DLdata <- imp_sim_list[,Xrep, drop = FALSE] colnames(DLdata) ### now 1:10 is the first simulation ### spilt into 100 simulations each by 10 save.image(file = "DL_nnet_scan_olden_sim1.RData") ###calculate q-values DLdata1=apply(DLdata,2,normalize) Simqvalue=DLqvalues(DLdata1,10) Loci<-rep("Neutral", 1000) Loci[c(1,11,21,31,41,51,61,71,81,91)]<-"QTL1" Loci[c(101,111,121,131,141,151,161,171,181,191)]<-"QTL2" Loci[c(201,211,221,231,241,251,261,271,281,291)]<-"QTL3" p.values<-data.frame(index = c(1:1000), DL_mlp=-log10(Simqvalue$q.values),Loci=Loci) Selected_Loci<-p.values$Loci[-which(p.values$Loci=="Neutral")]
Plotting Manhattan plot
p4 <- ggplot() + geom_point(aes(x=p.values$index[-which(p.values$Loci!="Neutral")], y=p.values$DL_mlp[-which(p.values$Loci!="Neutral")]), col = "gray83") + geom_point(aes(x=as.vector(p.values$index[-which(p.values$Loci=="Neutral")]), y=as.vector(p.values$DL_mlp[-which(p.values$Loci=="Neutral")]), colour = Selected_Loci)) + xlab("SNP (with maf > 0.05)") + ylab("DL_MLP -log10(p-values)") + ylim(0,1) + theme_bw() p4 #save.image(file = "DL_nnet_Scan_sim1_manhattan.RData")
Using other machine learning models
Besides the neural netwok model, other machine learning models also present impressive performance in various areas, such as extreme gradient boosting, random forest,etc. Now, we will try some extreme gradient boosting examples.
Extreme gradient boosting with “xgbTree”
econtrol <- caret::trainControl( ## 5-fold CV, repeat 5 times method = "adaptive_cv", number = 5, ## repeated ten times repeats = 5, adaptive = list(min = 5, alpha = 0.05,method = "gls", complete = TRUE),search = "random") ### xgbTree example for (i in 1:length(para)){ print(paste0("sim_1_",para[i],"tuning xboost tree 10 parameters")) Sys.time() #env=normalize(simdata[[1]][,2:11]) #genotype_norm=as.data.frame(apply(simdata,2,normalize)) # simf=as.formula(paste(colnames(env)[i],paste(names(genotype_norm[,15:1014]), collapse="+"),sep="~")) model_xgbTree_envi=caret::train(x=genotype, y=env[,i],### using genotype_norm probably will generate 0 values and the RMSE can get NA method="xgbTree", metric = "MAE",## "Accuracy", "RMSE" #preProcess=c("scale"), tuneLength = 10, ### search 100 combinations of parameters # verbose=1 is reporting the progress,o is sclience trControl = econtrol) print(paste0("sim_",1,"_",para[i],"tuning xgbTree finished")) Sys.time() save(model_xgbTree_envi,file=paste0("sim_",1,"_",para[i],"_xgbTree_Scan_env_trained_model.RData")) ### olden importance GradiexgbTree_simimp1=varImp(model_xgbTree_envi) locname=colnames(genotype) macha=match(locname,rownames(GradiexgbTree_simimp1$importance)) gbtreeimp=cbind(rownames(GradiexgbTree_simimp1$importance),GradiexgbTree_simimp1$importance) gbtreeimp1=gbtreeimp[macha,] write.csv(gbtreeimp1$Overall,file = paste0("sim_",1,"_",para[i],"_xgbTree_Scan_importance_env.csv")) write.csv(model_xgbTree_envi$results,file = paste0("sim_",1,"_",para[i],"_model_xgbTree_envi_tuning.csv") ) #registerDoSEQ() #stopCluster(cl) }
DLqvalues<-function(DL_data,K,estimation="auto") { require(robust) require(qvalue) loadings<-DL_data# [,1:as.numeric(K)] resscale <- apply(loadings, 2, scale) resmaha <- robust::covRob(resscale, distance = TRUE, na.action= na.omit, estim=estimation)$dist lambda <- median(resmaha)/qchisq(0.5,df=K) reschi2test <- stats::pchisq(resmaha/lambda,K,lower.tail=FALSE) qval <- qvalue::qvalue(reschi2test) q.values_DL<-qval$qvalues padj <- stats::p.adjust(reschi2test,method="bonferroni") return(data.frame(p.values=reschi2test, q.values=q.values_DL,padj=padj)) }
Reading importance values and calculate the p-values/q-values
impdir=list.dirs(path = ".", full.names = TRUE, recursive = TRUE) impfile <- list.files(impdir, pattern="*_xgbTree_Scan_importance_env.csv", full.names = TRUE) ## 100 simulations and 10 envir variables, impSim_all=lapply(impfile, read.csv) ### including all 100 simulations and 10 variables each simuilations, ### combine every factor (envir) into one file, meaning combining each single simulation into one file imp_sim_list=as.data.frame(do.call(cbind, impSim_all)) ### remove the loci ID Xrep<- !names(imp_sim_list) %in% c("X") DLdata <- imp_sim_list[,Xrep, drop = FALSE] colnames(DLdata) ### now 1:10 is the first simulation ### spilt into 100 simulations each by 10 save.image(file = "DL_xgbtree_scan_sim1.RData") ###calculate q-values DLdata1=apply(DLdata,2,normalize) Simqvalue=DLqvalues(DLdata1,10,estimation="auto") Loci<-rep("Neutral", 1000) Loci[c(1,11,21,31,41,51,61,71,81,91)]<-"QTL1" Loci[c(101,111,121,131,141,151,161,171,181,191)]<-"QTL2" Loci[c(201,211,221,231,241,251,261,271,281,291)]<-"QTL3" p.values<-data.frame(index = c(1:1000), DL_mlp=-log10(Simqvalue$q.values),Loci=Loci) Selected_Loci<-p.values$Loci[-which(p.values$Loci=="Neutral")]
Plotting Manhatan plot
p5 <- ggplot() + geom_point(aes(x=p.values$index[-which(p.values$Loci!="Neutral")], y=p.values$DL_mlp[-which(p.values$Loci!="Neutral")]), col = "gray83") + geom_point(aes(x=as.vector(p.values$index[-which(p.values$Loci=="Neutral")]), y=as.vector(p.values$DL_mlp[-which(p.values$Loci=="Neutral")]), colour = Selected_Loci)) + xlab("SNP (with maf > 0.05)") + ylab("DL_MLP -log10(p-values)") + ylim(0,1000) + theme_bw() p5 #save.image(file = "DL_xgbtree_Scan_sim1_manhattan.RData")
Extreme gradient boosting with “xgbDART”
econtrol <- caret::trainControl( ## 5-fold CV, repeat 5 times method = "adaptive_cv", number = 5, ## repeated ten times repeats = 5, adaptive = list(min = 5, alpha = 0.05,method = "gls", complete = TRUE),search = "random") ### xgbDART example for (i in 1:length(para)){ print(paste0("sim_1_",para[i],"tuning xboost tree 50 parameters")) Sys.time() #env=normalize(simdata[[1]][,2:11]) #genotype_norm=as.data.frame(apply(simdata,2,normalize)) # simf=as.formula(paste(colnames(env)[i],paste(names(genotype_norm[,15:1014]), collapse="+"),sep="~")) model_xgbDART_envi=caret::train(x=genotype, y=env[,i],### using genotype_norm probably will generate 0 values and the RMSE can get NA method="xgbDART", metric = "MAE",## "Accuracy", "RMSE" #preProcess=c("scale"), tuneLength = 50, ### search 100 combinations of parameters # verbose=1 is reporting the progress,o is sclience trControl = econtrol) print(paste0("sim_",1,"_",para[i],"tuning xgbDART finished")) Sys.time() save(model_xgbDART_envi,file=paste0("sim_",1,"_",para[i],"_xgbDART_Scan_env_trained_model.RData")) ### olden importance GradiexgbDART_simimp1=varImp(model_xgbDART_envi) locname=colnames(genotype) macha=match(locname,rownames(GradiexgbDART_simimp1$importance)) gbtreeimp=cbind(rownames(GradiexgbDART_simimp1$importance),GradiexgbDART_simimp1$importance) gbtreeimp1=gbtreeimp[macha,] write.csv(gbtreeimp1$Overall,file = paste0("sim_",1,"_",para[i],"_xgbDART_Scan_importance_env.csv")) write.csv(model_xgbDART_envi$results,file = paste0("sim_",1,"_",para[i],"_model_xgbDART_envi_tuning.csv") ) #registerDoSEQ() #stopCluster(cl) }
read importance values
impdir=list.dirs(path = ".", full.names = TRUE, recursive = TRUE) impfile <- list.files(impdir, pattern="*_xgbDART_Scan_importance_env.csv", full.names = TRUE) ## 100 simulations and 10 envir variables, impSim_all=lapply(impfile, read.csv) ### including all 100 simulations and 10 variables each simuilations, ### combine every factor (envir) into one file, meaning combining each single simulation into one file imp_sim_list=as.data.frame(do.call(cbind, impSim_all)) ### remove the loci ID Xrep<- !names(imp_sim_list) %in% c("X") DLdata <- imp_sim_list[,Xrep, drop = FALSE] colnames(DLdata) ### now 1:10 is the first simulation ### spilt into 100 simulations each by 10 save.image(file = "DL_xgbDART_scan_sim1.RData") ###calculate q-values DLdata1=apply(DLdata,2,normalize) Simqvalue=DLqvalues(DLdata1,10,estimation="auto") Loci<-rep("Neutral", 1000) Loci[c(1,11,21,31,41,51,61,71,81,91)]<-"QTL1" Loci[c(101,111,121,131,141,151,161,171,181,191)]<-"QTL2" Loci[c(201,211,221,231,241,251,261,271,281,291)]<-"QTL3" p.values<-data.frame(index = c(1:1000), DL_mlp=-log10(Simqvalue$q.values),Loci=Loci) Selected_Loci<-p.values$Loci[-which(p.values$Loci=="Neutral")]
Plotting Manhattan plot
load(file = "DL_xgbDART_Scan_sim1_manhattan.RData") p6 <- ggplot() + geom_point(aes(x=p.values$index[-which(p.values$Loci!="Neutral")], y=p.values$DL_mlp[-which(p.values$Loci!="Neutral")]), col = "gray83") + geom_point(aes(x=as.vector(p.values$index[-which(p.values$Loci=="Neutral")]), y=as.vector(p.values$DL_mlp[-which(p.values$Loci=="Neutral")]), colour = Selected_Loci)) + xlab("SNP (with maf > 0.05)") + ylab("DL_MLP -log10(p-values)") + ylim(0,150) + theme_bw() p6
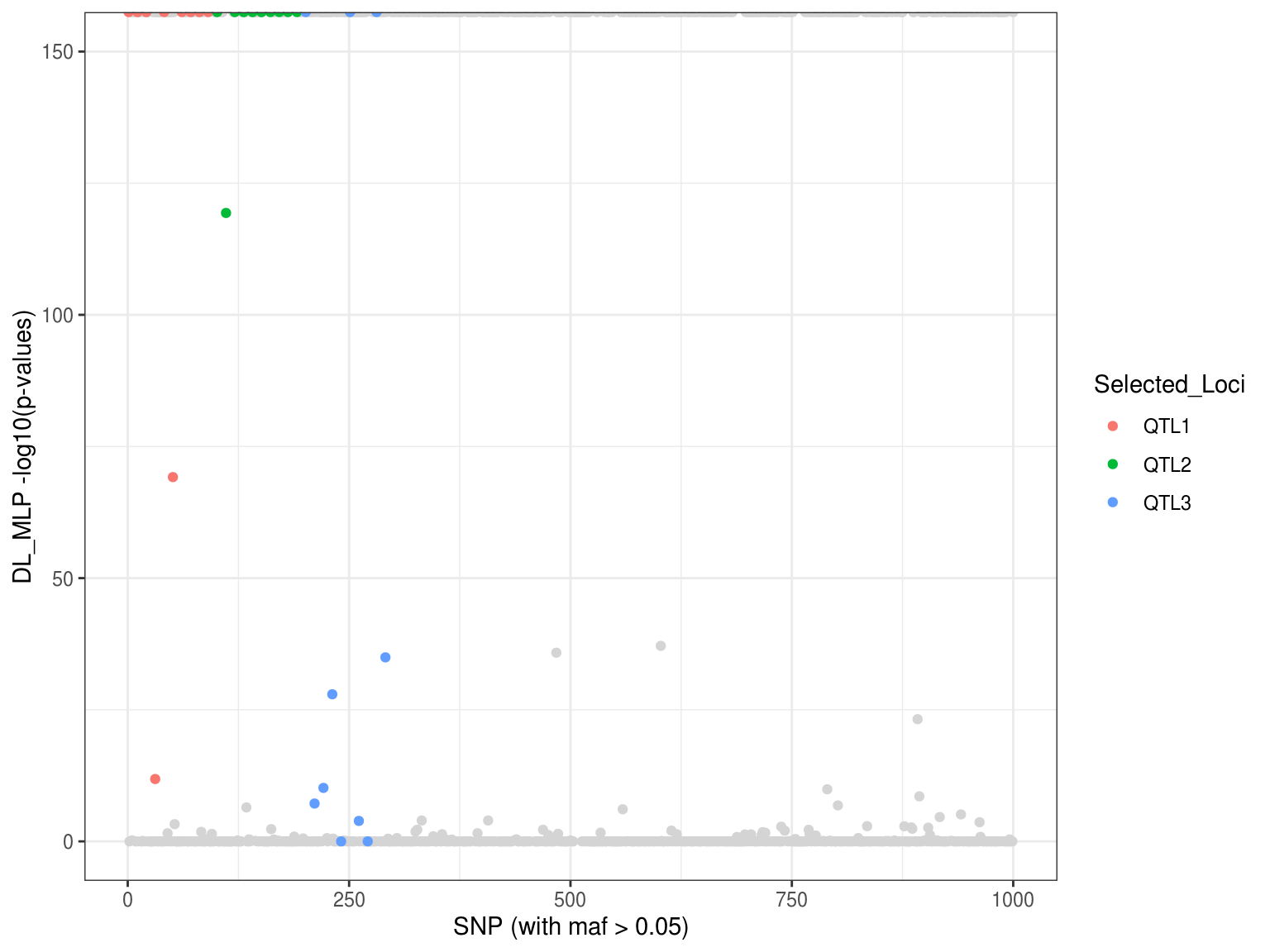
Note the outliers detection method based on mahalanobis distance and p-values no longer work here becasue the distances might not follow chi-square distribution. Please see the below section using autoencoder to correct the mahalanobis distance-based method.
Multi-Step Adaptive MCP-Net (msaenet) example
This method implements the multi-step adaptive elastic-net method introduced in Xiao and Xu (2015) for feature selection in high-dimensional regressions.
#Multi-Step Adaptive MCP-Net econtrol <- caret::trainControl( ## 5-fold CV, repeat 5 times method = "adaptive_cv", number = 5, ## repeated ten times repeats = 5, adaptive = list(min = 5, alpha = 0.05,method = "gls", complete = TRUE),search = "random") ### msaenet example for (i in 1:length(para)){ print(paste0("sim_1_",para[i],"tuning Multi-Step Adaptive MCP-Net tree 10 parameters")) Sys.time() #env=normalize(simdata[[1]][,2:11]) #genotype_norm=as.data.frame(apply(simdata,2,normalize)) # simf=as.formula(paste(colnames(env)[i],paste(names(genotype_norm[,15:1014]), collapse="+"),sep="~")) model_msaenet_envi=caret::train(x=genotype, y=env[,i],### using genotype_norm probably will generate 0 values and the RMSE can get NA method="msaenet", metric = "MAE",## "Accuracy", "RMSE" #preProcess=c("scale"), tuneLength = 10, ### search 100 combinations of parameters # verbose=1 is reporting the progress,o is sclience trControl = econtrol) print(paste0("sim_",1,"_",para[i],"tuning msaenet finished")) Sys.time() save(model_msaenet_envi,file=paste0("sim_",1,"_",para[i],"_msaenet_Scan_env_trained_model.RData")) ### olden importance msaenet_simimp1=varImp(model_msaenet_envi) locname=colnames(genotype) macha=match(locname,rownames(msaenet_simimp1$importance)) msaenetimp=as.data.frame(cbind(rownames(msaenet_simimp1$importance),msaenet_simimp1$importance)) msaenetimp1=msaenetimp[macha,] write.csv(msaenetimp1$Overall,file = paste0("sim_",1,"_",para[i],"_msaenet_Scan_importance_env.csv")) write.csv(model_msaenet_envi$results,file = paste0("sim_",1,"_",para[i],"_model_msaenet_envi_tuning.csv") ) #registerDoSEQ() #stopCluster(cl) }
Reading importance values
impdir=list.dirs(path = ".", full.names = TRUE, recursive = TRUE) impfile <- list.files(impdir, pattern="*_msaenet_Scan_importance_env.csv", full.names = TRUE) ## 100 simulations and 10 envir variables, impSim_all=lapply(impfile, read.csv) ### including all 100 simulations and 10 variables each simuilations, ### combine every factor (envir) into one file, meaning combining each single simulation into one file imp_sim_list=as.data.frame(do.call(cbind, impSim_all)) ### remove the loci ID Xrep<- !names(imp_sim_list) %in% c("X") DLdata <- imp_sim_list[,Xrep, drop = FALSE] colnames(DLdata) ### now 1:10 is the first simulation ### spilt into 100 simulations each by 10 save.image(file = "DL_msaenet_scan_sim1.RData") ###calculate q-values DLdata1=apply(DLdata,2,normalize) Simqvalue=DLqvalues(DLdata1,10,estimation="auto") Loci<-rep("Neutral", 1000) Loci[c(1,11,21,31,41,51,61,71,81,91)]<-"QTL1" Loci[c(101,111,121,131,141,151,161,171,181,191)]<-"QTL2" Loci[c(201,211,221,231,241,251,261,271,281,291)]<-"QTL3" p.values<-data.frame(index = c(1:1000), DL_mlp=-log10(Simqvalue$q.values),Loci=Loci) Selected_Loci<-p.values$Loci[-which(p.values$Loci=="Neutral")]
##load("DL_msaenet_Scan_sim1_manhattan.RData") p7 <- ggplot() + geom_point(aes(x=p.values$index[-which(p.values$Loci!="Neutral")], y=p.values$DL_mlp[-which(p.values$Loci!="Neutral")]), col = "gray83") + geom_point(aes(x=as.vector(p.values$index[-which(p.values$Loci=="Neutral")]), y=as.vector(p.values$DL_mlp[-which(p.values$Loci=="Neutral")]), colour = Selected_Loci)) + xlab("SNP (with maf > 0.05)") + ylab("DL_MLP -log10(p-values)") + ylim(0,150) + theme_bw() p7
Random forest example using “ranger”
## random forest ## Use GenABEL interface to read Plink data into R and grow a classification forest ## The ped and map files are not included library(GenABEL) convert.snp.ped("data.ped", "data.map", "data.raw") dat.gwaa <- load.gwaa.data("data.pheno", "data.raw") phdata(dat.gwaa)$trait <- factor(phdata(dat.gwaa)$trait) ranger(trait ~ ., data = dat.gwaa) for (i in 1:length(para)){ print(paste0("sim_1_",para[i]," random forest for SNP identification ")) Sys.time() #env=normalize(simdata[[1]][,2:11]) #genotype_norm=as.data.frame(apply(simdata,2,normalize)) # simf=as.formula(paste(colnames(env)[i],paste(names(genotype_norm[,15:1014]), collapse="+"),sep="~")) model_ranger_envi=caret::train(x=genotype, y=env[,i],### using genotype_norm probably will generate 0 values and the RMSE can get NA method="ranger", metric = "MAE",## "Accuracy", "RMSE" #preProcess=c("scale"), tuneLength = 10, ### search 100 combinations of parameters # verbose=1 is reporting the progress,o is sclience trControl = econtrol,importance="impurity_corrected",replace = FALSE) print(paste0("sim_",1,"_",para[i],"tuning ranger finished")) Sys.time() save(model_ranger_envi,file=paste0("sim_",1,"_",para[i],"_ranger_Scan_env_trained_model.RData")) ### olden importance ranger_simimp1=caret::varImp(model_ranger_envi) write.csv(ranger_simimp1$importance,file = paste0("sim_",1,"_",para[i],"_ranger_Scan_importance_env.csv")) write.csv(model_ranger_envi$results,file = paste0("sim_",1,"_",para[i],"_model_ranger_envi_tuning.csv") ) }
impdir=list.dirs(path = "/home/xinghu2019/Simulations/DeepGenomeScan_v0.1/vignettes", full.names = TRUE, recursive = TRUE)
impfile <- list.files(impdir, pattern="*_ranger_Scan_importance_env.csv", full.names = TRUE)
## 100 simulations and 10 envir variables,
impSim_all=lapply(impfile, read.csv) ### including all 100 simulations and 10 variables each simuilations,
### combine every factor (envir) into one file, meaning combining each single simulation into one file
imp_sim_list=as.data.frame(do.call(cbind, impSim_all))
### remove the loci ID
Xrep<- !names(imp_sim_list) %in% c("X")
DLdata <- imp_sim_list[,Xrep, drop = FALSE]
colnames(DLdata) ### now 1:10 is the first simulation
### spilt into 100 simulations each by 10
save.image(file = "DL_ranger_scan_sim1.RData")
###calculate q-values
mahalanobis1=function (x, center, cov, inverted = FALSE, ...)
{
x <- if (is.vector(x))
matrix(x, ncol = length(x))
else as.matrix(x)
if (!isFALSE(center))
x <- sweep(x, 2L, center)
if (!inverted)
cov <- solve(cov, tol=1e-500,...)
setNames(rowSums(x %*% cov * x), rownames(x))
}
covRob=function (data, corr = FALSE, distance = TRUE, na.action = na.fail,
estim = "auto", control = covRob.control(estim, ...), ...)
{
data <- na.action(data)
if (is.data.frame(data))
data <- data.matrix(data)
n <- nrow(data)
p <- ncol(data)
rowNames <- dimnames(data)[[1]]
colNames <- dimnames(data)[[2]]
dimnames(data) <- NULL
if (is.null(colNames))
colNames <- paste("V", 1:p, sep = "")
if (p < 2)
stop(sQuote("data"), " must have at least two columns to compute ",
"a covariance matrix")
if (n < p)
stop("not enough observations")
estim <- casefold(estim)
if (estim == "auto") {
if ((n < 1000 && p < 10) || (n < 5000 && p < 5))
estim <- "donostah"
else if (n < 50000 && p < 20)
estim <- "mcd"
else estim <- "pairwiseqc"
control <- covRob.control(estim)
}
else {
dots <- list(...)
dots.names <- names(dots)
if (any(dots.names == "quan") && all(dots.names != "alpha")) {
dots.names[dots.names == "quan"] <- "alpha"
names(dots) <- dots.names
}
if (any(dots.names == "ntrial") && all(dots.names !=
"nsamp")) {
dots.names[dots.names == "ntrial"] <- "nsamp"
names(dots) <- dots.names
}
control.names <- names(control)
if (any(control.names == "init.control"))
control.names <- c(control.names, names(control$init.control))
if (any(!is.element(dots.names, control.names))) {
bad.args <- sQuote(setdiff(dots.names, control.names))
if (length(bad.args) == 1)
stop(sQuote(bad.args), " is not a control argument for the ",
dQuote(estim), " estimator")
else stop(paste(sQuote(bad.args), collapse = ", "),
" are not control ", "arguments for the ", dQuote(estim),
" estimator")
}
}
ans <- switch(estim, donostah = {
args <- list(x = data)
if (control$nresamp != "auto") args$nsamp <- control$nresamp
if (control$maxres != "auto") args$maxres <- control$maxres
if (!control$random.sample) set.seed(21)
args$tune <- control$tune
args$prob <- control$prob
args$eps <- control$eps
ds <- do.call("CovSde", args)
list(cov = getCov(ds), center = getCenter(ds), dist = getDistance(ds))
}, pairwiseqc = {
x <- CovOgk(data, control = CovControlOgk(smrob = "s_mad",
svrob = "qc"))
list(center = getCenter(x), cov = getCov(x), dist = getDistance(x),
raw.center = x@raw.center, raw.cov = x@raw.cov, raw.dist = x@raw.mah)
}, pairwisegk = {
x <- CovOgk(data)
list(center = getCenter(x), cov = getCov(x), dist = getDistance(x),
raw.center = x@raw.center, raw.cov = x@raw.cov, raw.dist = x@raw.mah)
}, m = {
mcd.control <- control$init.control
control$init.control <- NULL
if (mcd.control$alpha > 1) mcd.control$alpha <- mcd.control$alpha/n
init <- robustbase::covMcd(data, cor = FALSE, control = mcd.control)
ans <- rrcov::covMest(data, cor = FALSE, r = control$r, arp = control$arp,
eps = control$eps, maxiter = control$maxiter, t0 = init$raw.center,
S0 = init$raw.cov)
ans$dist <- ans$mah
ans$raw.center <- init$raw.center
ans$raw.cov <- init$raw.cov
ans$raw.dist <- init$raw.mah
ans
}, mcd = {
if (control$alpha > 1) control$alpha <- control$alpha/n
ans <- robustbase::covMcd(data, cor = FALSE, control = control)
ans$center <- ans$raw.center
ans$cov <- ans$raw.cov
ans$dist <- ans$raw.mah
ans$raw.cov <- ans$raw.cov/prod(ans$raw.cnp2)
ans$raw.dist <- ans$raw.mah * prod(ans$raw.cnp2)
ans
}, weighted = {
if (control$alpha > 1) control$alpha <- control$alpha/n
ans <- robustbase::covMcd(data, cor = FALSE, control = control)
ans$dist <- ans$mah
ans$raw.cov <- ans$raw.cov/prod(ans$raw.cnp2)
ans$raw.dist <- ans$raw.mah * prod(ans$raw.cnp2)
ans
}, default = stop("Invalid choice of estimator."))
dimnames(ans$cov) <- list(colNames, colNames)
names(ans$center) <- colNames
if (is.null(ans$raw.cov)) {
ans$raw.cov <- NA
ans$raw.center <- NA
}
else {
dimnames(ans$raw.cov) <- list(colNames, colNames)
names(ans$raw.center) <- colNames
}
if (distance) {
if (is.null(ans$dist))
ans$dist <- mahalanobis1(data, ans$center, ans$cov)
if (!is.na(ans$raw.cov[1])) {
if (is.null(ans$raw.dist))
ans$raw.dist <- mahalanobis(data, ans$raw.center,
ans$raw.cov)
}
else ans$raw.dist <- NA
}
else {
ans$dist <- NA
ans$raw.dist <- NA
}
if (!is.na(ans$dist[1]) && !is.null(rowNames))
names(ans$dist) <- rowNames
if (!is.na(ans$raw.dist[1]) && !is.null(rowNames))
names(ans$raw.dist) <- rowNames
if (corr) {
std <- sqrt(diag(ans$cov))
ans$cov <- ans$cov/(std %o% std)
if (!is.na(ans$raw.cov[1])) {
std <- sqrt(diag(ans$raw.cov))
ans$raw.cov <- ans$raw.cov/(std %o% std)
}
}
ans$corr <- corr
ans$estim <- estim
ans$control <- control
ans$call <- match.call()
ans <- ans[c("call", "cov", "center", "dist", "raw.cov",
"raw.center", "raw.dist", "corr", "estim", "control")]
oldClass(ans) <- "covRob"
ans
}
DLqvalues<-function(DL_data,K,estimation="auto")
{
require(robust)
require(qvalue)
loadings<-DL_data# [,1:as.numeric(K)]
resscale <- apply(loadings, 2, scale)
resmaha <-covRob(resscale, distance = TRUE, na.action= na.omit, estim=estimation)$dist
lambda <- median(resmaha)/qchisq(0.5,df=K)
reschi2test <- stats::pchisq(resmaha/lambda,K,lower.tail=FALSE)
qval <- qvalue::qvalue(reschi2test)
q.values_DL<-qval$qvalues
padj <- stats::p.adjust(reschi2test,method="bonferroni")
return(data.frame(p.values=reschi2test, q.values=q.values_DL,padj=padj))
}
DLdata1=apply(DLdata,2,normalize)
Simqvalue=DLqvalues(DLdata1[,-c(4,7)],8,estimation="auto") ### the discrete environmnet confound the True discovery
#### Try autoencoder to construct the outlier detection
library(h2o)
#
# ----------------------------------------------------------------------
#
# Your next step is to start H2O:
# > h2o.init()
#
# For H2O package documentation, ask for help:
# > ??h2o
#
# After starting H2O, you can use the Web UI at http://localhost:54321
# For more information visit https://docs.h2o.ai
#
# ----------------------------------------------------------------------
#
# Attaching package: 'h2o'
# The following objects are masked from 'package:stats':
#
# cor, sd, var
# The following objects are masked from 'package:base':
#
# ||, &&, %*%, apply, as.factor, as.numeric, colnames,
# colnames<-, ifelse, %in%, is.character, is.factor, is.numeric,
# log, log10, log1p, log2, round, signif, trunc
localH2O = h2o.init()
Loimp<-as.h2o(DLdata, destination_frame="train.hex")
feature_names=colnames(DLdata)
prostate.dl = h2o.deeplearning(x = feature_names, training_frame = Loimp,
autoencoder = TRUE,
reproducible = T,
seed = 1234,
hidden = c(10,10,10), epochs = 1000)
# Locimpout = h2o.anomaly(DLdata, Loimp, per_feature=TRUE)
# head(Locimpout)
Locimpout = h2o.anomaly(prostate.dl, Loimp, per_feature=FALSE)
head(Locimpout)
err <- as.data.frame(Locimpout)
prostate_anon = h2o.anomaly(prostate.dl, Loimp)
head(prostate_anon)
prostate_anon_per_feature = h2o.anomaly(prostate.dl, Loimp, per_feature = TRUE)
head(prostate_anon_per_feature)
Loci<-rep("Neutral", 1000)
Loci[c(1,11,21,31,41,51,61,71,81,91)]<-"QTL1"
Loci[c(101,111,121,131,141,151,161,171,181,191)]<-"QTL2"
Loci[c(201,211,221,231,241,251,261,271,281,291)]<-"QTL3"
p.values<-data.frame(index = c(1:1000), DL_mlp=-log10(Simqvalue$q.values),Loci=Loci)
Selected_Loci<-p.values$Loci[-which(p.values$Loci=="Neutral")]
MLP_MSE<-data.frame(index = c(1:1000), DL_mlp=(err$Reconstruction.MSE),Loci=Loci)
Selected_Loci<-p.values$Loci[-which(p.values$Loci=="Neutral")]
# [1] "Overall" "Overall.1" "Overall.2" "Overall.3" "Overall.4"
# [6] "Overall.5" "Overall.6" "Overall.7" "Overall.8" "Overall.9"
# Connection successful!
#
# R is connected to the H2O cluster:
# H2O cluster uptime: 4 hours 6 minutes
# H2O cluster timezone: Europe/London
# H2O data parsing timezone: UTC
# H2O cluster version: 3.30.1.3
# H2O cluster version age: 7 months and 4 days !!!
# H2O cluster name: H2O_started_from_R_root_bki425
# H2O cluster total nodes: 1
# H2O cluster total memory: 1.56 GB
# H2O cluster total cores: 4
# H2O cluster allowed cores: 4
# H2O cluster healthy: TRUE
# H2O Connection ip: localhost
# H2O Connection port: 54321
# H2O Connection proxy: NA
# H2O Internal Security: FALSE
# H2O API Extensions: Amazon S3, XGBoost, Algos, AutoML, Core V3, TargetEncoder, Core V4
# R Version: R version 3.6.1 (2019-07-05)
#
#
|
| | 0%
|
|=================================================================| 100%
#
|
| | 0%
|
|==== | 6%
|
|======== | 12%
|
|============ | 18%
|
|=============== | 23%
|
|================== | 28%
|
|====================== | 33%
|
|========================= | 38%
|
|============================ | 42%
|
|============================== | 47%
|
|================================= | 51%
|
|==================================== | 55%
|
|======================================= | 60%
|
|========================================= | 64%
|
|============================================ | 67%
|
|============================================== | 71%
|
|================================================= | 75%
|
|==================================================== | 79%
|
|====================================================== | 83%
|
|======================================================== | 86%
|
|========================================================== | 90%
|
|============================================================= | 94%
|
|=============================================================== | 97%
|
|=================================================================| 100%
# Reconstruction.MSE
# 1 4.944123e-04
# 2 1.014526e-04
# 3 1.197233e-04
# 4 5.173995e-04
# 5 2.302771e-05
# 6 1.088714e-04
# Reconstruction.MSE
# 1 4.944123e-04
# 2 1.014526e-04
# 3 1.197233e-04
# 4 5.173995e-04
# 5 2.302771e-05
# 6 1.088714e-04
# reconstr_Overall.SE reconstr_Overall.1.SE reconstr_Overall.2.SE
# 1 7.514726e-05 1.992749e-03 1.376523e-05
# 2 1.516515e-07 4.758491e-05 3.185421e-05
# 3 2.396642e-05 1.159243e-05 5.209207e-05
# 4 1.297110e-04 4.532196e-05 6.378183e-05
# 5 1.077169e-04 5.117244e-07 4.623367e-06
# 6 5.721887e-06 6.060704e-05 1.683548e-05
# reconstr_Overall.3.SE reconstr_Overall.4.SE reconstr_Overall.5.SE
# 1 4.236208e-04 1.927254e-03 5.590005e-05
# 2 6.385523e-04 3.786654e-05 1.424314e-05
# 3 4.340652e-04 7.868405e-10 3.627963e-05
# 4 5.867697e-06 4.163324e-06 2.677203e-05
# 5 5.867697e-06 3.554956e-06 1.561003e-05
# 6 6.515638e-04 2.681937e-05 9.315670e-06
# reconstr_Overall.6.SE reconstr_Overall.7.SE reconstr_Overall.8.SE
# 1 4.228555e-06 2.856027e-04 7.640090e-06
# 2 4.228555e-06 1.196248e-04 4.665664e-05
# 3 4.228555e-06 5.589126e-04 2.433334e-05
# 4 4.228555e-06 4.865315e-03 2.842759e-05
# 5 4.228555e-06 7.560494e-05 6.107906e-06
# 6 2.831970e-04 3.153456e-05 5.536286e-07
# reconstr_Overall.9.SE
# 1 1.582160e-04
# 2 7.376275e-05
# 3 5.176155e-05
# 4 4.065183e-07
# 5 6.451005e-06
# 6 2.566018e-06Outlier detection using autoencoder
This section will demonstrate how to use the autoencoder to detect the outliers when using p-values or q-values do not work (see example in )
library(ggplot2) p8 <- ggplot() + geom_point(aes(x=MLP_MSE$index[-which(MLP_MSE$Loci!="Neutral")], y=MLP_MSE$DL_mlp[-which(MLP_MSE$Loci!="Neutral")]), col = "gray83") + geom_point(aes(x=as.vector(MLP_MSE$index[-which(MLP_MSE$Loci=="Neutral")]), y=as.vector(MLP_MSE$DL_mlp[-which(MLP_MSE$Loci=="Neutral")]), colour = Selected_Loci)) + xlab("SNP (with maf > 0.05)") + ylab("DL_MLP_MSE") + ylim(0,0.4) + theme_bw() p8
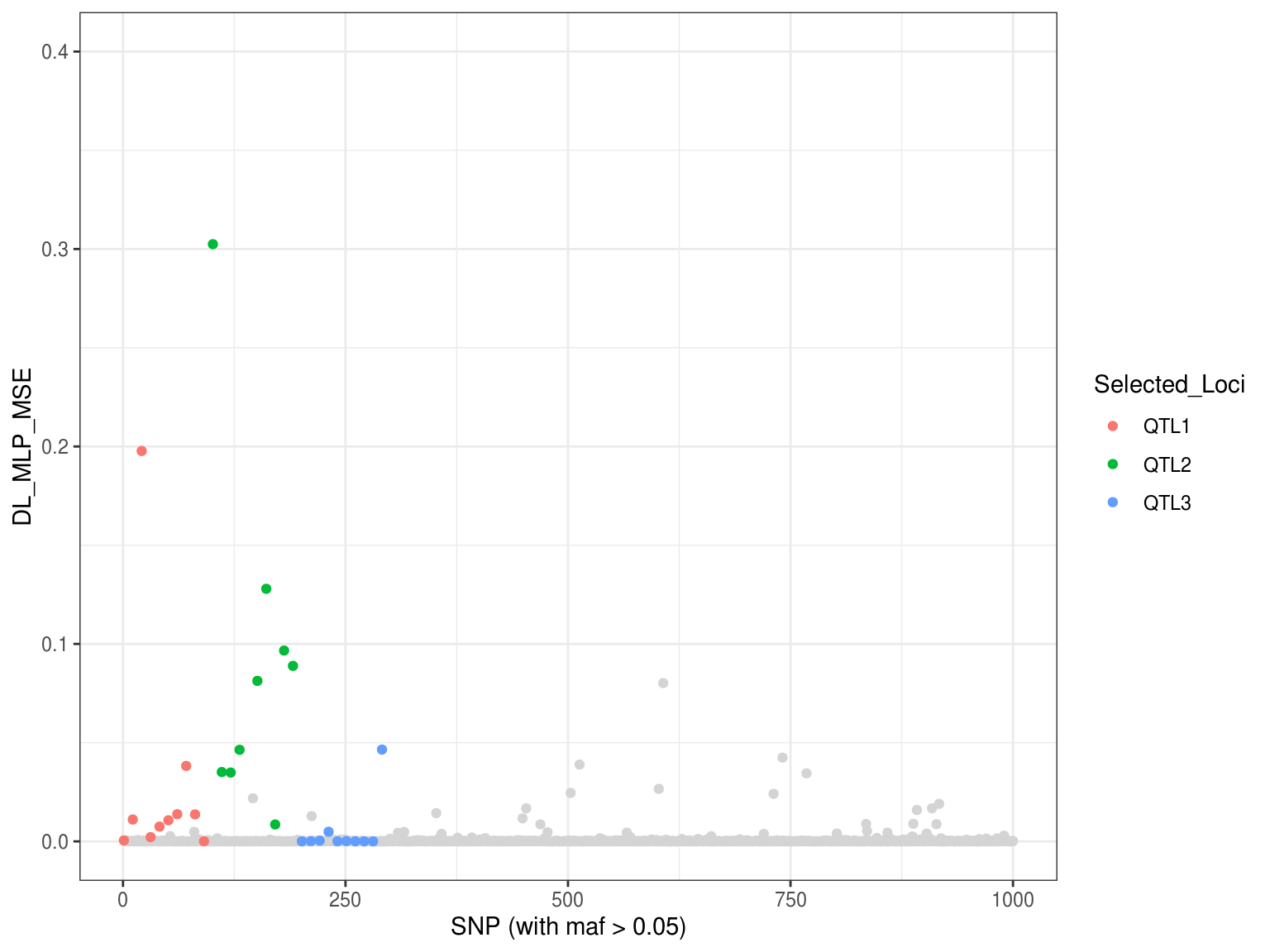
##save.image(file = "DL_ranger_Scan_sim1_manhattan.RData")Now the MSE-based Manhattan plot seems better compared to the p-values/q-values above.
We will post and update the outlier method in DeepGenomeScan in our next version. We also need to test its performance compared to p-values/q-values.
Neural network MLP example using “neuralnet”
########## neuralnet mlp random search############################################ set.seed(123) econtrol <- caret::trainControl(## 5-fold CV, repeat 5 times method = "adaptive_cv", number = 5, ## repeated ten times repeats = 5, adaptive = list(min = 5, alpha = 0.05,method = "gls", complete = TRUE), search = "random" ) softplus <- function(x) log(1 + exp(x)) ### now we use self-defined smooth RELU function softplus <- function(x) log(1 + exp(x)) ### indeed this function learns well with old version of neuralnet package. return to the older version if necessary which will have less hyperparameter (difficult to overfit with older version) ### mlpneuralnet # #### mlpneuralnet1 k=2 a=0.8 linear=function(x) a*x customRelu =function(x) {x/(1+exp(-2*k*x))} softplus =function(x) log(1 + exp(x)) devtools::install_github("bips-hb/neuralnet") #### Note this architecture has whether the output activatioin is linear, some studies say linear is more effective for unbounded regression, when linear output is TRUE, the activation 2 is invalid ### some time the simpler the faster and the better #### The developement version of neuralnet, which allows specifying the output function mlpneuralnet1<- list(label = "Neural Network", library = "neuralnet", loop = NULL, type = c('Regression'), parameters = data.frame(parameter = c('layer1', 'layer2', 'layer3',"activation1","activation2","linear.output"), class = c('numeric', 'numeric', 'numeric',"character","character","character"), label = c('Number of hidden Units in Layer 1', 'number of hidden Units in Layer 2', 'number of hidden Units in Layer 3',"Activation function in hidden layer","Activation function in output layer","Activation function linear out choice")), grid = function(x, y, len = NULL, search = "grid") { afuncs=c("logistic", "tanh","softplus") outputf=c("TRUE","FALSE") if(search == "grid") { out <- expand.grid(layer1 = ((1:len) * 2) - 1, layer2 = 0, layer3 = 0, activation1=c("logistic", "tanh","softplus"),activation2=c("logistic", "tanh","softplus"),linear.output=c("TRUE","FALSE")) } else { out <- data.frame(layer1 = sample(2:20, replace = TRUE, size = len), layer2 = sample(c(0, 2:20), replace = TRUE, size = len), layer3 = sample(c(0, 2:20), replace = TRUE, size = len), activation1=sample(afuncs, size = len, replace = TRUE), activation2=sample(afuncs, size = len, replace = TRUE), linear.output=sample(outputf,size = len,replace = TRUE)) } out }, fit = function(x, y, wts, param, lev, last, classProbs, ...) { colNames <- colnames(x) dat <- if(is.data.frame(x)) x else as.data.frame(x, stringsAsFactors = TRUE) dat$.outcome <- y form <- as.formula(paste(".outcome ~",paste(colNames, collapse = "+"))) if(param$layer1 == 0) stop("the first layer must have at least one hidden unit") if(param$layer2 == 0 & param$layer2 > 0) stop("the second layer must have at least one hidden unit if a third layer is specified") nodes <- c(param$layer1) if(param$layer2 > 0) { nodes <- c(nodes, param$layer2) if(param$layer3 > 0) nodes <- c(nodes, param$layer3) } actf=(param$activation1)### note the difference in c(param$activation) for this model and other model, becaue the self-defined softplus function can't be a vector, so here we should not use c(,softplus) outputactf=(param$activation2) linear.output=c(param$linear.output) neuralnet::neuralnet(form, algorithm="rprop+",data = dat,rep=1, hidden = nodes, stepmax = 1e+09, learningrate.factor = list(minus = 0.5,plus = 1.2),act.fct=actf,output.act.fct=outputactf,linear.output=linear.output,...) }, predict = function(modelFit, newdata, submodels = NULL) { newdata <- newdata[, modelFit$model.list$variables, drop = FALSE] predict(modelFit, covariate = newdata)$net.result[,1] }, varImp = function(object, ...){ imps <- NeuralNetTools::olden(object,bar_plot =FALSE) out <- data.frame(Overall = as.vector(imps)) rownames(out) <- names(imps) out }, prob = NULL, tags = c("Neural Network"), sort = function(x) x[order(x$layer1, x$layer2, x$layer3,x$activation1,x$activation2,x$linear.output),]) for (i in 1:length(para)){ print(paste0("sim_1_",para[i]," train neuralnet")) Sys.time() #env=normalize(simdata[[j]][,2:11]) genotype_norm=as.data.frame(apply(sim_example,2,normalize)) simf=as.formula(paste(colnames(env)[i],paste(names(genotype_norm[,15:1014]), collapse="+"),sep="~")) model_neuralnet_envi_mlp=DeepGenomeScan(simf, data=as.data.frame(genotype_norm), method="neuralnet", metric = "RMSE",## "Accuracy", "RMSE" #preProcess=c("scale"), tuneLength = 10, ### search 10 combinations of parameters # verbose=1 is reporting the progress,o is sclience trControl = econtrol) print(paste0("sim_1_",para[i],"tuning neuralnet finished")) Sys.time() save(model_neuralnet_envi_mlp,file=paste0("sim_1_",para[i],"_mlpneuralnet_env_trained_model.RData")) ### olden importance neuralnet_simimp1=NeuralNetTools::olden(model_neuralnet_envi_mlp$finalModel, bar_plot=FALSE) write.csv(neuralnet_simimp1,file = paste0("sim_1_",para[i],"_mlpneuralnet_importance_env.csv")) write.csv(model_neuralnet_envi_mlp$results,file = paste0("sim_1_",para[i],"_mlpneuralnet_env_tuning.csv") ) print(paste0("sim_1_",para[i],"importance output finished")) #registerDoSEQ() #stopCluster(cl) } #save.image(file = "neuralnet_mlp_Scan_sims_final_after_trained_data.RData") DLqvalues<-function(DL_data,K,estimation="auto") { require(robust) require(qvalue) loadings<-DL_data# [,1:as.numeric(K)] resscale <- apply(loadings, 2, scale) resmaha <- robust::covRob(resscale, distance = TRUE, na.action= na.omit, estim=estimation)$dist lambda <- median(resmaha)/qchisq(0.5,df=K) reschi2test <- stats::pchisq(resmaha/lambda,K,lower.tail=FALSE) qval <- qvalue::qvalue(reschi2test) q.values_DL<-qval$qvalues padj <- stats::p.adjust(reschi2test,method="bonferroni") return(data.frame(p.values=reschi2test, q.values=q.values_DL,padj=padj)) }
impdir=list.dirs(path = ".", full.names = TRUE, recursive = TRUE) impfile <- list.files(impdir, pattern="*_mlpneuralnet_importance_env.csv", full.names = TRUE) ## 100 simulations and 10 envir variables, impSim_all=lapply(impfile, read.csv) ### including all 100 simulations and 10 variables each simuilations, ### combine every factor (envir) into one file, meaning combining each single simulation into one file imp_sim_list=as.data.frame(do.call(cbind, impSim_all)) ### remove the loci ID Xrep<- !names(imp_sim_list) %in% c("X") DLdata <- imp_sim_list[,Xrep, drop = FALSE] colnames(DLdata) ### now 1:10 is the first simulation ### spilt into 100 simulations each by 10 save.image(file = "DL_mlp_neural_Scan_sim1.RData") ###calculate q-values DLdata1=apply(DLdata,2,normalize) Simqvalue=DLqvalues(DLdata1,10) Loci<-rep("Neutral", 1000) Loci[c(1,11,21,31,41,51,61,71,81,91)]<-"QTL1" Loci[c(101,111,121,131,141,151,161,171,181,191)]<-"QTL2" Loci[c(201,211,221,231,241,251,261,271,281,291)]<-"QTL3" p.values<-data.frame(index = c(1:1000), DL_mlp=-log10(Simqvalue$p.values),Loci=Loci) Selected_Loci<-p.values$Loci[-which(p.values$Loci=="Neutral")]
Manhattan plot
p10 <- ggplot() + geom_point(aes(x=p.values$index[-which(p.values$Loci!="Neutral")], y=p.values$DL_mlp[-which(p.values$Loci!="Neutral")]), col = "gray83") + geom_point(aes(x=as.vector(p.values$index[-which(p.values$Loci=="Neutral")]), y=as.vector(p.values$DL_mlp[-which(p.values$Loci=="Neutral")]), colour = Selected_Loci)) + xlab("SNP (with maf > 0.05)") + ylab("DL_MLP -log10(p-values)") + ylim(0,20) + theme_bw() p10
MLP model implementation with default seting. Details about the ready-to-use model in DeepGenomeScan framework can be found here:https://xinghuq.github.io/DeepGenomeScan/articles/DeepGenomeScan_model_list.html
### using the available model in DeepGenomeScan mlp_scan<-caret::train(x=as.matrix(geno_impu),y=(klfdapcmat[,1]), method="mlp", metric = "MAE",## "Accuracy", "RMSE","Rsquared","MAE" tuneLength = 10, ### 11 tunable parameters 11^2 # , ### or search 100 combinations of parameters using random tuneLength=100 verbose=0,# verbose=1 is reporting the progress,o is sclience trControl = econtrol1,importance = FALSE) for (i in 1:length(klfdapcmat)){ mlp_scan1<- caret::train(geno_norm,klfdapcmat[,i], method="mlp", metric = "RMSE",## "Accuracy", "RMSE","Rsquared","MAE" tuneLength = 50, ### 11 tunable parameters 11^2 # , ### or search 100 combinations of parameters using random tuneLength=100 verbose=0,# verbose=1 is reporting the progress,o is sclience trControl = econtrol1,importance = FALSE) Sys.time() save(mlp_scan1,file = paste0("sim1_",colnames(klfdapcmat)[i],"_mlpRSNNSDecay_klfdapcmat_Model.RData")) RSNNS_simimp1=olden(mlp_scan1$finalModel,bar_plot=FALSE) save(RSNNS_simimp1,file = paste0("sim1_",colnames(klfdapcmat)[i],"_mlpRSNNSDecay_klfdapcmat_importance.RData")) write.csv(RSNNS_simimp1$importance,file = paste0("sim_",colnames(klfdapcmat)[i],"_mlpRSNNSDecay_imp.csv")) write.csv(mlp_scan1$results,file = paste0("sim_",colnames(klfdapcmat)[i],"_model_RSNNSDecay_klfdapcmati_tuning.csv")) } Sys.time() save.image(file = "mlp_scan_klfdapcmat1_sim_test_Final.RData")
Calculating p-values/q-values and plot Manhattan plot
Loci<-rep("Neutral", 1000) Loci[c(1,11,21,31,41,51,61,71,81,91)]<-"QTL1" Loci[c(101,111,121,131,141,151,161,171,181,191)]<-"QTL2" Loci[c(201,211,221,231,241,251,261,271,281,291)]<-"QTL3" scaled_imp=normalizeData(RSNNS_simimp1$importance,type = "0_1") SNPimp<-data.frame(index = c(1:1000), MLP= -log10(scaled_imp),Loci=Loci) Selected_Loci=SNPimp$Loci[-which(SNPimp$Loci=="Neutral")] p2 <- ggplot() + geom_point(aes(x=SNPimp$index[-which(SNPimp$Loci!="Neutral")], y=SNPimp$MLP[-which(SNPimp$Loci!="Neutral")]), col = "gray83") + geom_point(aes(x=as.vector(SNPimp$index[-which(SNPimp$Loci=="Neutral")]), y=as.vector(SNPimp$MLP[-which(SNPimp$Loci=="Neutral")]), colour = Selected_Loci)) + xlab("SNP (with maf > 0.05)") + ylab("MLP -log10(q-values)") + ylim(0,10) + theme_bw() p2
Let’s build and train a deep learning modoel using h2o package, which also provide several other good algorthms such as glmnet, Gradient Boosting Machines.
# The following two commands remove any previously installed H2O packages for R. if ("package:h2o" %in% search()) { detach("package:h2o", unload=TRUE) } if ("h2o" %in% rownames(installed.packages())) { remove.packages("h2o") } # Next, we download packages that H2O depends on. pkgs <- c("RCurl","jsonlite") for (pkg in pkgs) { if (! (pkg %in% rownames(installed.packages()))) { install.packages(pkg) } } # Now we download, install and initialize the H2O package for R. install.packages("h2o", type="source", repos="https://h2o-release.s3.amazonaws.com/h2o/rel-zeno/3/R") # Finally, let's load H2O and start up an H2O cluster library(h2o) h2o.init() library("h2o") #start h2o localH2o <- h2o.init(nthreads = -1, max_mem_size = "6G")
Gernerally, this approach takes longer time than adaptive cross-validation.
Compiling DL_h2o in DeepGenomeScan framework, for GUP version, please use H2O4GPU package. Note that different libraries have different data format/processing.
econtrol1 <- trainControl(## 5-fold CV, repeat 5 times method = "adaptive_cv", number = 5, ## repeated ten times repeats = 5,search = "random") set.seed(999) options(warn=-1) model_h2o_mlp<- caret::train(as.matrix(genotype_norm),env$envir1, method=DL_h2o, metric = "RMSE",## "Accuracy", "RMSE","Rsquared","MAE" tuneLength = 10, ### 11 tunable parameters 11^2 # , ### or search 100 combinations of parameters using random tuneLength=100 trControl = econtrol1) #### There is a model specific varIMP for this model varImp(model_h2o_mlp,scale = FALSE) out <- as.data.frame(h2o::h2o.varimp(model_h2o_mlp$finalModel), stringsAsFactors = TRUE) colnames(out)[colnames(out) == "scaled_importance"] <- "Overall" rownames(out) <- out$names save.image(file = "example_tutorial_Data.RData")
Compiling CNNsgd
CNN_sgd <- list(label = "Convolutional Neural Network", library = "keras", loop = NULL, type = c('Regression', "Classification"), parameters = data.frame( parameter = c("nFilter","nStride",'lambda', "units1","units2","dropout", "activation1","activation2","activation3"), class = c(rep('numeric', 6), "character","character","character"), label = c("no. of convolutions","stride between convolutions",'L2 Regularization', "hidden_neurons1","hidden_neurons2", "drop_out_rates","Activation Function convol layer","Activation function dense hidden layer","Activation function layer to output") ), grid = function(x, y, len = NULL, search = "grid") { afuncs <- c("sigmoid", "relu", "tanh","linear") if(search == "grid") { out <- expand.grid(nFilter=as.integer(nrow(x)/10),nStride=3, lambda = c(0, 10 ^ seq(-1, -4, length = len - 1)), units1 = ((1:len) * 2) - 1, units2 = ((1:len) * 1) - 1, dropout = seq(0, .5, length = len), activation1 = "relu",activation2="sigmoid",activation3="linear" ) } else { n <- nrow(x) out <- data.frame(nFilter=sample(10:500, replace = TRUE, size = len),nStride=sample(2:20, replace = TRUE, size = len), lambda = 10^runif(len, min = -5, 1),units1 = sample(2:n*2, replace = TRUE, size = len),units2 = sample(1:as.integer(n/2)-1, replace = TRUE, size = len), dropout = runif(len, max = .5),activation1 = sample(afuncs, size = len, replace = TRUE),activation2 = sample(afuncs, size = len, replace = TRUE), activation3 = sample(afuncs, size = len, replace = TRUE)) } out }, fit = function(x, y, wts, last,lev,param, ...) { require(dplyr) K <- keras::backend() K$clear_session() # if(!is.matrix(x)) x <- as.matrix(x) ########## ### here should reshape the data################################# Note: the key step feed to the cnn x=kerasR::expand_dims(x,axis=2) ################ ###### also this define the shape of the tensor###### argument: input_shape input_shape = c(nSNP,1) nSNP=dim(x)[2] model<-keras::keras_model_sequential() %>% keras::layer_conv_1d(filters = param$nFilter, kernel_size = c(3), strides=param$nStride,activation =as.character(param$activation1), input_shape = c(nSNP,1),kernel_regularizer = keras::regularizer_l2(param$lambda)) %>% layer_max_pooling_1d(pool_size = c( 2)) %>% # add pooling layer: takes maximum of two consecutive values layer_flatten() %>% layer_dropout(rate=param$dropout) %>% layer_dense(units = param$units1, activation =as.character(param$activation2)) %>% layer_dense(units = param$units2, activation =as.character(param$activation3)) # model<-keras::keras_model_sequential() %>% # layer_conv_1d(filters = param$nFilter, kernel_size = c(3), strides=param$nStride,activation =as.character(param$activation1), # input_shape = c(1000,1),kernel_regularizer = keras::regularizer_l2(param$lambda)) %>% # layer_max_pooling_1d(pool_size = c( 2)) %>% # add pooling layer: takes maximum of two consecutive values # layer_flatten() %>% # layer_dropout(rate=param$dropout) %>% # layer_dense(units = param$units1, activation =as.character(param$activation2)) %>% # layer_dense(units = param$units2, activation =as.character(param$activation3))%>% #### activation function of last layer # layer_dense(units = 1) ### this should be the same as the input shape: input_shape = c(nSNP,1) #more complex model # model<-keras::keras_model_sequential() %>% # keras::layer_conv_1d(filters=128, kernel_size = 3, strides=3,activation =as.character(param$activation1), # input_shape = c(nSNPs, 1),kernel_regularizer = keras::regularizer_l2(param$lambda)) %>% # layer_max_pooling_1d(pool_size = 2) %>% # layer_conv_1d(filters = 64, kernel_size =3, activation = as.character(param$activation1)) %>% # layer_flatten() %>% # layer_dropout(rate=param$dropout) %>% # layer_dense(units =param$units, activation = as.character(param$activation2)) #Compile the model. Note that this linked above %>% # model%>% compile( # optimizer='sgd', # loss='mean_squared_error', # metrics='mean_squared_error') if(is.factor(y)) { y <- class2ind(y) model %>% layer_dense(units = 1) %>% #### activation function of last layer keras::compile( loss = "categorical_crossentropy", optimizer = "sgd", metrics = "accuracy" ) } else { model %>% layer_dense(units = 1) %>% #### activation function of last layer keras::compile( loss = "mean_squared_error", optimizer = "sgd", metrics = "mean_squared_error" ) } model %>% keras::fit( x = x, y = y, kernel_regularizer = keras::regularizer_l2(param$lambda), ... ) if(last) model <- keras::serialize_model(model) list(object = model) }, predict = function(modelFit, newdata, submodels = NULL) { if(inherits(modelFit, "raw")) modelFit$object <- keras::unserialize_model(modelFit) # if(!is.matrix(newdata)) # newdata <- as.matrix(newdata) newdata=kerasR::expand_dims(newdata,axis=2) out <- keras::predict_on_batch(modelFit$object, newdata) ## check for model type if(ncol(out) == 1) { out <- out[, 1] } else { out <- modelFit$obsLevels[apply(out, 1, which.max)] } out }, prob = function(modelFit, newdata, submodels = NULL) { if(inherits(modelFit, "raw")) modelFit$object <- keras::unserialize_model(modelFit) # if(!is.matrix(newdata)) #newdata <- as.matrix(newdata) newdata=kerasR::expand_dims(newdata,axis=2) out <- keras::predict_on_batch(modelFit$object, newdata) colnames(out) <- modelFit$obsLevels as.data.frame(out, stringsAsFactors = TRUE) }, varImp = NULL,### CNN importance will estimate separately tags = c(" CNN", "L2 Regularization"), sort = function(x) x[order(x$nFilter, -x$lambda),], notes = paste("After `train` completes, the keras model object is serialized", "so that it can be used between R session. When predicting, the", "code will temporarily unsearalize the object. To make the", "predictions more efficient, the user might want to use ", "`keras::unsearlize_model(object$finalModel$object)` in the current", "R session so that that operation is only done once.", "Also, this model cannot be run in parallel due to", "the nature of how tensorflow does the computations.", "Unlike other packages used by `train`, the `dplyr`", "package is fully loaded when this model is used."), check = function(pkg) { testmod <- try(keras::keras_model_sequential(), silent = TRUE) if(inherits(testmod, "try-error")) stop("Could not start a sequential model. ", "`tensorflow` might not be installed. ", "See `?install_tensorflow`.", call. = FALSE) TRUE }) econtrol1 <- trainControl(## 5-fold CV, repeat 5 times method = "repeatedcv", number = 5, ## repeated ten times repeats = 5,search = "random") genotype_norm=as.data.frame(apply(genotype,2,normalize)) model_Keras_CNN<- DeepGenomeScan(x=(genotype_norm),y=(env$envir1), method=CNNsgd, metric = "MAE",## "Accuracy", "RMSE","Rsquared" tuneLength = 10, ### 11 tunable parameters 11^2 # , ### or search 100 combinations of parameters using random tuneLength=100 verbose=0,# verbose=1 is reporting the progress,o is sclience trControl = econtrol1,importance = FALSE)
permutation varimp importance
optmodel=keras::unserialize_model(model_Keras_CNN$finalModel$object) optmodel pred = keras::predict_on_batch(optmodel, as.matrix(genotype_norm)) set.seed(123) feature_names=colnames(genotype) library(doParallel) cl <- makePSOCKcluster(5) registerDoParallel(cl) Tensor_sim_imp_cnn=CNN_varIMP_permute(optmodel, feature_names, train_y=env$envir1, train_x=as.matrix(genotype_norm), #smaller_is_better = FALSE, type = c("difference", "ratio"), nsim = 10,## MCMC permutation large numbers need much more time sample_size = NULL, sample_frac = NULL, verbose = FALSE, progress = "none", parallel = TRUE, paropts = NULL) Tensor_sim_imp_NULL_cnn=CNN_varIMP_NULL_model(optmodel, feature_names, train_y=env$envir1, train_x=as.matrix(genotype_norm), #smaller_is_better = FALSE, type = c("difference", "ratio"), nsim = 10,## MCMC permutation large numbers need much more time sample_size = NULL, sample_frac = NULL, verbose = FALSE, progress = "none", parallel = TRUE, paropts = NULL) registerDoSEQ() stopCluster(cl) VarImps_permcnn=Tensor_sim_imp_cnn$CNN_Decrease_acc VarImps_nullcnn=Tensor_sim_imp_NULL_cnn$CNN_Decrease_acc save.image(file = "CNN_Sim1_env1_test.RData")
The above tutorial shows some machine learning, especially deep learning based method for detecting natural selection. The basic step is to construct a model first and compile it with DeepGenomeScan framework, then do genome scan to calculate the SNP importance. Finally we use the importance values to identify the outliers as the candidate loci under selection.
Some examples run very long time, so we did not show all the results, users can implement these methods on your own machine.
Please email qinxinghu@gmail.com if you have any questions when you use this package and if you found bugs.
Welcome contribute more frameworks and models to DeepGenomeScan.
Reference
R. A. Maronna and R. H. Zamar (2002) Robust estimates of location and dispersion of high-dimensional datasets. Technometrics 44 (4), 307–317.
Wang, J., Zamar, R., Marazzi, A., Yohai, V., Salibian-Barrera, M., Maronna, R., … & Maechler, M. (2017). robust: Port of the S+“Robust Library”. R package version 0.4-18. Available online at: https://CRAN. R-project. org/package= robust.
Abadi, M., Barham, P., Chen, J., Chen, Z., Davis, A., Dean, J., … & Kudlur, M. (2016). Tensorflow: A system for large-scale machine learning. In 12th {USENIX} symposium on operating systems design and implementation ({OSDI} 16) (pp. 265-283).
Bergmeir, C. N., & Benítez Sánchez, J. M. (2012). Neural networks in R using the Stuttgart neural network simulator: RSNNS. American Statistical Association.
Beck, M. W. (2018). NeuralNetTools: Visualization and analysis tools for neural networks. Journal of statistical software, 85(11), 1.
Candel, A., Parmar, V., LeDell, E., & Arora, A. (2016). Deep learning with H2O. H2O. ai Inc.
Deane-Mayer, Z. A., & Knowles, J. E. (2016). caretEnsemble: ensembles of caret models. R package version, 2(0).
Gulli, A., & Pal, S. (2017). Deep learning with Keras. Packt Publishing Ltd.
Klima, G. (2016). FCNN4R: Fast Compressed Neural Networks for R. R package version 0.6, 2.
Kuhn, M. (2015). Caret: classification and regression training. ascl, ascl-1505.
Gevrey, M., Dimopoulos, I., & Lek, S. (2003). Review and comparison of methods to study the contribution of variables in artificial neural network models. Ecological modelling, 160(3), 249-264.
Xiao, Nan, and Qing-Song Xu. 2015. “Multi-Step Adaptive Elastic-Net: Reducing False Positives in High-Dimensional Variable Selection.” Journal of Statistical Computation and Simulation 85 (18): 3755–65.